In this study, the authors successfully constructed a microengineered HCC organoid chip based on an MSC-PDO-PBMC co-culture model to simulate HCC tumor microenvironment (TME) and enhance the efficacyand homogeneity of organoid culture. More importantly, this microfluidic platform significantly shortens the timeframe for high-throughput organoid culture and drug screening. It also more accurately predicts the response of clinical HCC patients to common anticancer drugs, particularly immunotherapy agents like ICIs. Consequently, it demonstrates tremendous potential in personalized cancer treatment and immunotherapy prognosis prediction.
In May 2023, Zhengyu Zou published a first-author paper titled “Micro-Engineered Organoid-on-a-Chip Based on Mesenchymal Stromal Cells to Predict Immunotherapy Responses of HCC Patients” in Advanced Science. The authors designed a multilayer microfluidic chip enabling high-throughput co-culture of mesenchymal stem cells, peripheral blood mononuclear cells, and patient-derived organoids. This approach promotes hepatocellular carcinoma organoid growth while simulating the tumor microenvironment by supporting monocyte/macrophage survival and M2 macrophage differentiation. The platform facilitates drug screening and immunotherapy response assessment, demonstrating significant potential for personalized cancer treatment and immunotherapy prognosis prediction.
Hepatocellular carcinoma (HCC) ranks as the sixth most common cancer globally and the third leading cause of cancer-related deaths. Immune checkpoint inhibitors (ICI) are increasingly used in HCC treatment, though thepopulation of beneficiaries remains limited. Patient selection for ICI therapy primarily relies on high PD-L1 expression and lymphocyte infiltration. However, the absence of the native tumor microenvironment (TME)—comprising immune cells and stromal cells such as cancer-associated fibroblasts (CAFs) and mesenchymal stromal cells—often leads to inaccurate prognosis prediction, as the TME significantly influences tumor progression and host response to immunotherapy, frequently leading to inaccurate prognosis prediction. Therefore, establishing an efficient drug screening platform that accounts for the TME—particularly tumor-associated macrophages—holds significant importance for personalized cancer therapy. Patient-derived organoids (PDOs), as 3D culture models capable of proliferating in vitro while retaining key characteristics of primary tumor specimens, also offer broad application prospects in drug screening and precision medicine. The potential of organoids to predict patient clinical outcomes remains unclear, and in vitro evaluation models for immunotherapy drugs are still lacking. Combined with existing challenges in culture techniques, these factors limit their application in personalized drug screening. In this study, the authors established an MSC-PDO-PBMC co-culture system. This system enhances the success rate and growth velocity of hepatocellular carcinoma organoid culture by supporting monocyte/macrophage survival and TAM differentiation, thereby mimicking the tumor microenvironment. Furthermore, we designed a microfluidic chip to enhance the homogeneity of high-throughput MSC-PDO-PBMC co-culture and enable dynamic drug delivery. This microengineered organoid chip utilizes engineered micropores and channels to refine the tumor engineering model simulating the TME. It not only substantially reduces the time and cost of HCC organoid culture and high-throughput drug screening but also demonstrates more accurate predictive potential in evaluating immunotherapy responses in HCC patients, thereby providing an efficient platform for personalized drug screening.
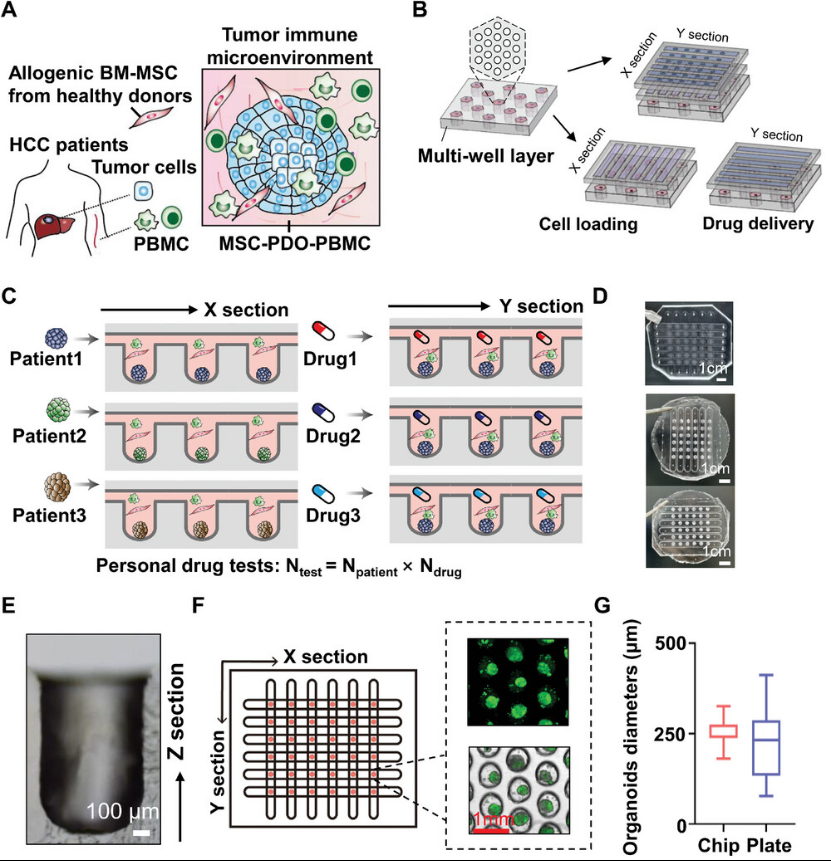
First, the authors co-cultured HCC tumor cells obtained via needle biopsy or surgical resection with autologous PBMCs from healthy donors and allogeneic bone marrow-derived MSCs (BM-MSCs) to reconstruct the TME (Figure 1). A microfluidic chip designed for high-throughput organoid culture and drug screening features top and middle layers dedicated to drug delivery (Y-segment) and cell loading (X-segment), each containing six vertically intersecting microchannels. The bottom layer for organoid culture and drug testing comprises 36 microarray units, each with 19 microwells arranged hexagonally for drug testing. Mixed MSC-PBMC-PDO can be loaded through the middle layer and trapped in the bottom layer micropores via turbulent flow stress, where they form HCC organoids simulating the TME. Drugs are then dynamically delivered from the top layer channels. This microchip enables high-throughput PDO culture and personalized drug testing. Optimized cell loading conditions ensure uniform PDO distribution across all microarray units with rapid organoid formation. Representative brightfield and fluorescent staining images of PDOs cultured on the microarray chip demonstrate high organoid homogeneity. HCC organoids cultured on the microchip for 7 days were then observed under microscopy. The size variation of PDOs cultured within the microchip (as indicated by PDO diameter dispersion) was significantly lower than that observed in conventional 96-well plate cultures. Co-cultured PDOs exhibited similar cellular composition patterns, demonstrating that the microchip substantially enhances the homogeneity of high-throughput PDO culture.
Figure 1
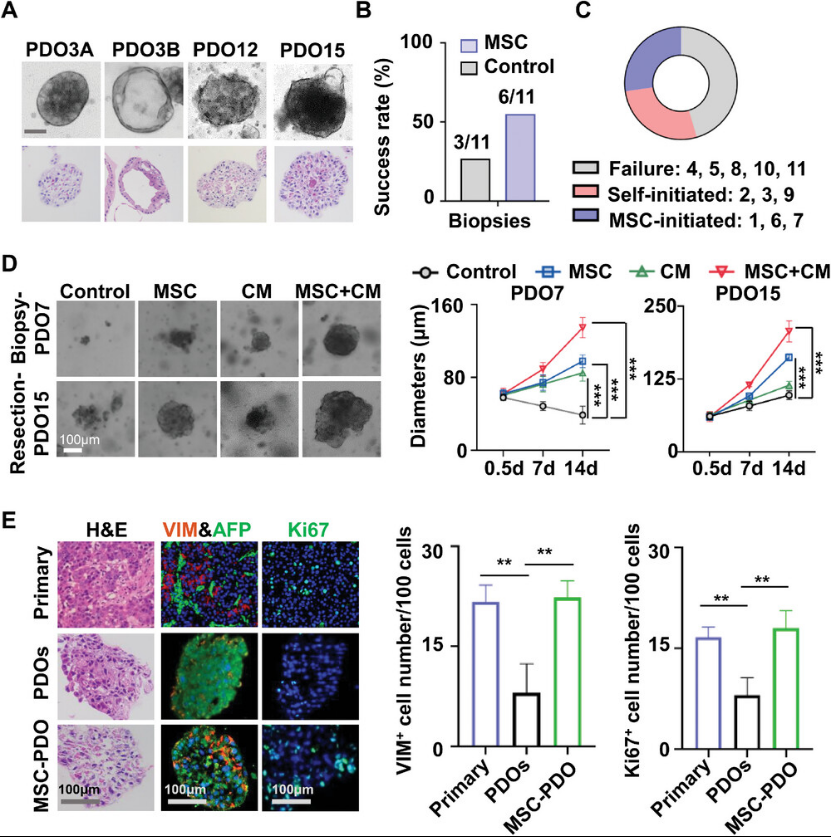
Next, we investigated the effects of MSCs on PDO culture from primary HCC specimens. Twelve PDOs were successfully established from 22 samples, with seven achieving stable establishment. MSCs significantly improved the success rate of PDO culture from biopsy-derived samples. During the culture process, the authors observed that co-culture with MSCs markedly accelerated PDO growth. Notably, the authors found that even in basal medium, the conditioned medium (CM) promoted tumor organoid growth without the need for supplemental specific cytokines. Hematoxylin and eosin (H&E) and immunofluorescence staining revealed that primary HCC tissue expressed alpha-fetoprotein (AFP, a HCC marker) and vimentin (VIM, a stromal cell marker), along with high expression of Ki67 (a cell proliferation marker). These characteristics were also observed in PDOs co-cultured with MSCs (Figure 2).
Figure 2
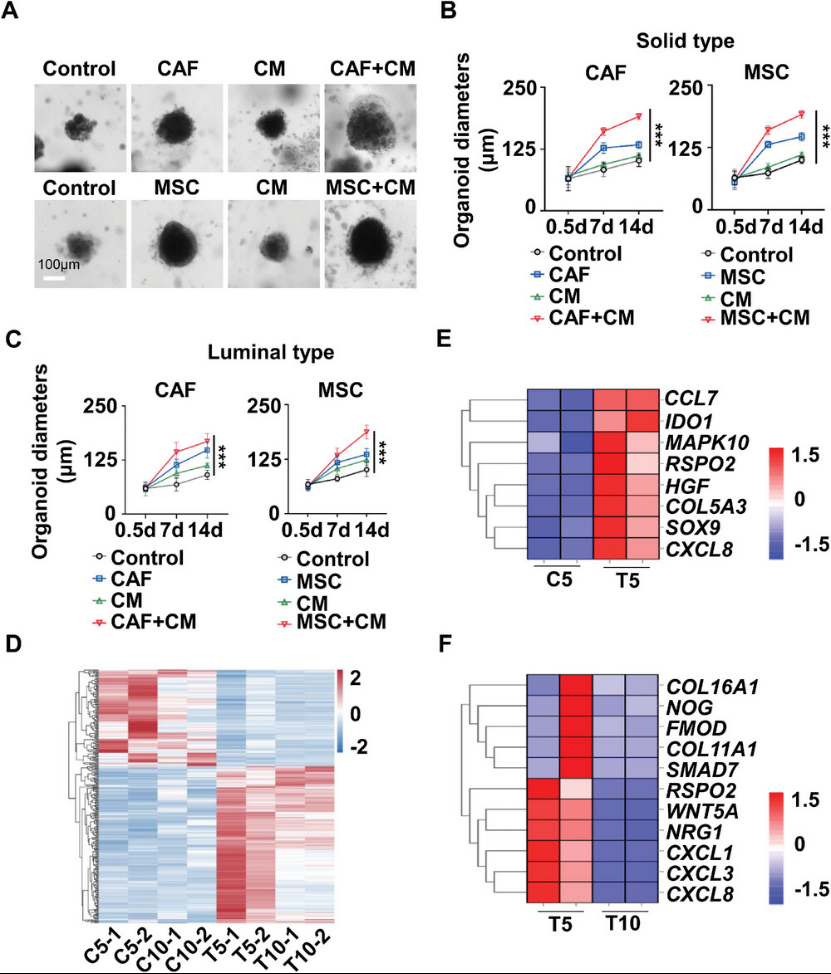
The authors isolated CAFs from HCC resection specimens, which exhibited morphology and phenotypes similar to BM-MSCs. When co-cultured with PDO15, both allogeneic MSCs and autologous CAFs demonstrated comparable efficacy in supporting the growth of HCC PDO-derived organoids. To assess whether MSCs could differentiate into CAFs within the HCC microenvironment and whether passage number affected MSC plasticity, the authors co-cultured MSCs at different passage levels with PDOs. They found that P10 MSCs promoted PDO growth more effectively than MSCs at higher passage levels. Treatment of P5 or P10 MSCs with PDO supernatant generated tumor-trained MSCs (TA-MSCs). RNA-seq analysis compared gene expression profiles between TA-MSCs and untreated MSCs. P5 MSCs, rather than P10 MSCs, exhibited a robust tumor-training response (Figure3), indicating upregulation of genes involved in tumor growth, chemotaxis, and immunosuppression. RT-PCR further validated the gene expression patterns of CAFs relative to MSCs from P5 or P10 passages. MSCs from the P5 passage exhibited a CAF-like phenotype after tumor education, whereas MSCs from the P10 passage did not, suggesting that MSCs progressively lose their differentiation capacity after 10 passages.
Figure 3
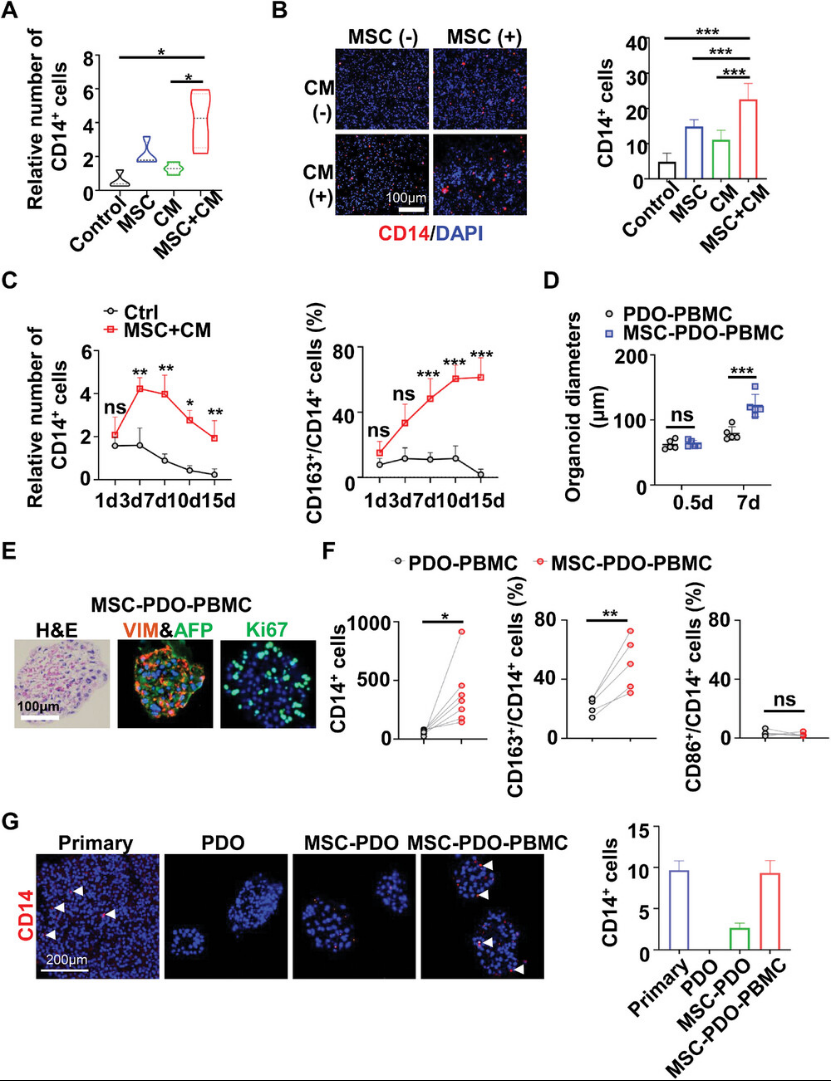
Monocytes/macrophages, as the predominant cell type in the tumor immune microenvironment, are prone to loss during PDO culture, while stromal cells play a crucial role in regulating their survival and differentiation. To investigate the role of mesenchymal stem cells in the long-term in vitro culture of monocytes/macrophages, the authors co-cultured PBMCs with MSCs at different ratios and analyzed the results via flow cytometry or immunostaining. MSCs or their conditioned media (CM) increased monocyte numbers in a ratio- or dose-dependent manner. MSCs and CM demonstrated synergistic effects in promoting monocyte/macrophage survival (Figure 4). MSC+CM significantly promoted monocyte proliferation within 7 days and sustained survival rates for 15 days, whereas macrophage colony-stimulating factor (M-CSF) supplementation marginally promoted monocyte proliferation within 3 days but failed to maintain monocyte survival beyond 3 days of culture (Figure 4C). MSC+CM treatment enhanced monocyte differentiation toward CD163 M2 macrophages but did not enhance CD86 M1 macrophage differentiation, with a slight reduction in CD3 T lymphocyte numbers following IL-2 stimulation. PCR and Western blot data further confirmed MSC-promoted M2 differentiation. These findings indicate that MSCs favor macrophage differentiation toward a TAM-like (M2) phenotype.
Figure 4
To generate PDO models simulating the TME of primary tumors, the authors co-cultured HCC PDOs with autologous PBMCs and MSCs at an optimized ratio. Consistent with observations in the MSC-PDO co-culture system, MSCs promoted PDO growth in the presence of autologous PBMCs (Figure 4). Immunohistochemical data revealed that HCC organoids generated from the MSC-PDO-PBMC model highly expressed AFP, VIM, and Ki67 (Figure 4). These features were detected in primary HCC tissue but absent in conventionally cultured PDOs (Figure 2). Furthermore, flow cytometric analysis demonstrated that MSCs significantly promoted monocyte survival and M2 differentiation in PDO-PBMC co-cultures without affecting M1 polarization (Figure 4). CD14+ macrophages were detected in organoids co-cultured with MSCs via immunostaining, while organoids cultured with MSCs plus PBMCs exhibited abundant macrophages similar to those in primary HCC tissue (Figure 4), indicating that the MSC-PDO-PBMC co-culture generated organoids resembling the native HCC TME.
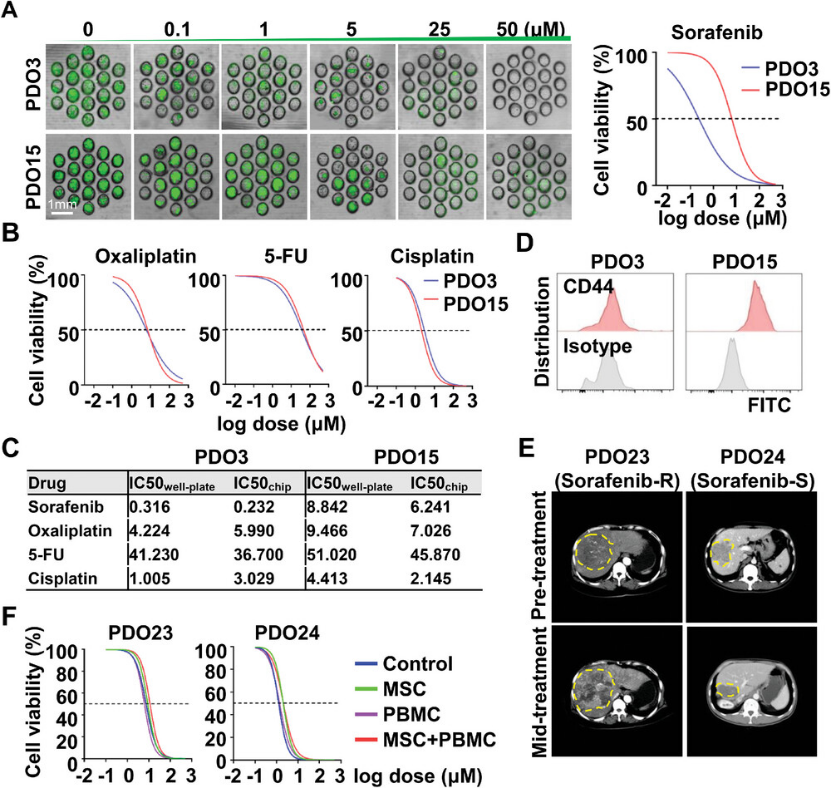
To evaluate model suitability for personalized drug screening, the authors treated MSC-PDO-PBMC and CAF-PDO-PBMC with sorafenib, oxaliplatin, 5-fluorouracil, and cisplatin, comparing results to conventionally cultured PDO models. Cell viability was assessed via CCK-8 assay, and dose-response curves were plotted to determine the half-maximal inhibitory concentration (IC₅₀). PDO3 and PDO15 exhibited distinct drug responses to sorafenib but similar responses to oxaliplatin, 5-FU, and cisplatin. Co-culture with allogeneic MSCs or autologous CAFs did not alter PDO sensitivity to these chemotherapeutic or targeted agents. To further evaluate the predictive accuracy of PDO-based drug sensitivity testing, previously cryopreserved biopsy-derived PDOs from HCC patients with differing sorafenib responses (PDO23: sorafenib-resistant; PDO24: sorafenib-sensitive) were evaluated for drug sensitivity with and without MSC/PBMC co-culture systems. The results aligned with the corresponding patient responses (Figure 5), indicating the significant clinical application potential of the drug screening microchips.
Figure 5
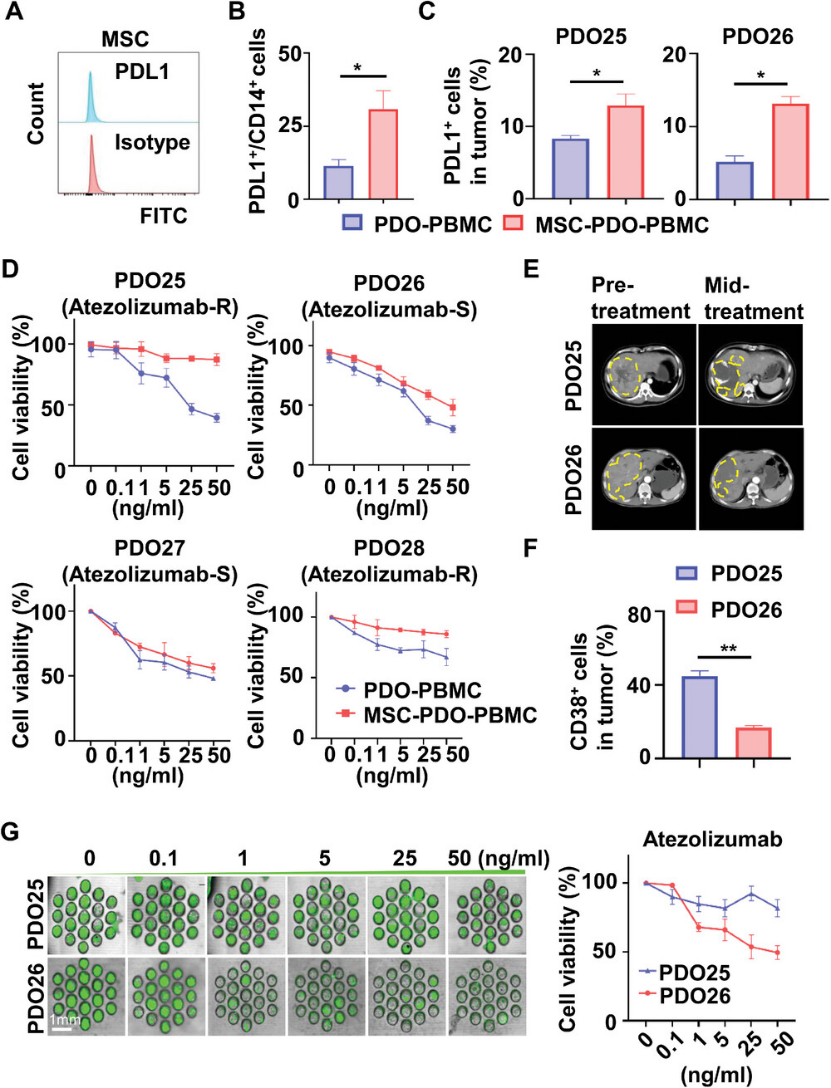
Finally, the authors evaluated the immunotherapy drug sensitivity of the PDO co-culture model. Normally expanded MSCs do not express PD-L1 (Figure 6), but PD-L1 expression increases in monocytes/macrophages and tumor cells during co-culture (Figure 6). Immunotherapy sensitivity was assessed using MSC-PDO-PBMC models constructed from HCC patients with differing responses to atezolizumab (anti-PD-L1 antibody) (Figure 6). Based on clinical information, patients PDO25 and PDO28 experienced disease progression after atezolizumab treatment, while patients PDO26 and PDO27 achieved partial remission (Figure 6). High CD38 expression in tumors is often associated with resistance to anti-PD-1/PD-L1 immunotherapy. Flow cytometry data showed significantly higher CD38 expression in PDO25 compared to PDO26 (Figure 6), while their PD-L1 expression was comparable, consistent with their clinical responses to atezolizumab. Drug sensitivity to atezolizumab was tested in PDO25 and PDO26 using different co-culture models in 96-well plates. For PDO25 (atezolizumab-resistant), both MSC-PDO-PBMC and CAF-PDO-PBMC models exhibited a resistant phenotype, whereas the PDO-PBMC model showed a sensitive response. In contrast, for PDO26 (anti-PD-L1-sensitive), the MSC-PDO-PBMC, CAF-PDO-PBMC, and PDO-PBMC models exhibited sensitive responses to anti-PD-L1 (Figure 6). Collectively, these data indicate that the MSC (CAF)-PDO-PBMC model, which mimics the native TME, is more suitable than the traditional PDO model for in vitro testing of immunotherapeutic drugs. Drug sensitivity testing conducted on microchips based on MSC-PDO-PBMC co-culture yielded results consistent with CCK-8 assays in 96-well plates (Figure 6). This microengineered organoid chip simulating the TME reduces organoid culture time to within one week, providing an efficient, high-throughput platform for immunotherapy drug screening.
Figure 6
In summary, in this study, the authors successfully constructed a microengineered HCC organoid chip based on an MSC-PDO-PBMC co-culture model to simulate the HCC tumor microenvironment (TME) and enhance the efficiency and homogeneity of organoid culture. More importantly, this microfluidic platform significantly shortens the timeframe for high-throughput organoid culture and drug screening while more accurately predicting clinical HCC patients' responses to common anticancer drugs, particularly immunotherapy agents like ICIs. Consequently, it demonstrates tremendous potential for personalized cancer treatment and immunotherapy prognosis prediction.